
seizure
-
متلازمة جولدنهار
متلازمة جولدنهار Goldenhar syndrome لهذه المتلازمة عدة أسماء .فيطلق عليها أيضا سلسلة(متلازمة) العين و الأذن و الفقرات(Oculo-Auriculo-Vertebrali Spectrum) كما…
أكمل القراءة » -
متلازمة الخيشوم والأذن والكلية
متلازمة الخيشوم والأذن والكلية Branchio-oto-renal syndrome و صف الدكتور ملنيك هذه المتلازمة في عام 1975…
أكمل القراءة » -
متلازمة اُوشر
متلازمة اُوشر Usher Syndrome يصيب هذا المرض جميع الشعوب و لكن الدكتور البرشت فون جريفي(. Albrecht Von Graefe) وصفة…
أكمل القراءة » -
شق الشفة و شق الحنك
شق الشفة و شق الحنك المرأة إذا رأت أرنبا فإن طفلها سيولد بشفة أرنبية!! في القديم كان هناك نظرة…
أكمل القراءة » -
الخلع الوركي الولادي
الخلع الوركي الولادي يعتقد الكثير خطاءً أن سبب خلع الورك الولادي لحديثي الولادة ناتج عن الآلات المستخدمة في التوليد أثناء…
أكمل القراءة » -
مقدمة عن المشاكل الخلقية
المشاكل الخلقية هي عبارة عن تخلق غير طبيعي في احد أعضاء الجسم أو الأنسجة في مرحلة تخلق الجنين. وعادة ما…
أكمل القراءة » -
الحثل العضلي الولادي
الحثل العضلي الولادي Congenital Muscular Dystrophy ما هو الحثل العضلي الولادي ؟ الحثل العضلي الولادي عباره عن مجموعه من الامراض…
أكمل القراءة » -
مرض الحثل العضلي
مقدمة عن مرض الحثل العضلي Muscular Dystrophy مرض الحِثل العضلي(Muscular Dystrophy) و يرمز له بالرمز MD هو احد امراض…
أكمل القراءة » -
مرض التليف العصبي
مرض التليف العصبي Neurofibromatosis هناك نوعان من التليف العصبي النوع الأول والنوع الثاني ومع أن لهم نفس المسمى إلا أنهما…
أكمل القراءة » -
التصلب الدرني الوراثي
التصلب الدرني الوراثي Tuberous Sclerosis التصلب الدرني هو خلل وراثي ينتقل لثلث الحالات المصابة عن طريق الوالدين اما باقي الحالات…
أكمل القراءة » -
متلازمة استطالة كيو تي
متلازمة استطالة كيو تي Long QT syndrome ماهي متلازمة استطالة كيو تي؟ إن متلازمة استطالة كيو تي هي مجموعة من…
أكمل القراءة » -
متلازمة إدوارد
Edward syndrome نزل متلازمة ادورد كملف بي دي اف للطباعة متلازمة إدوارد متلازمة إدوارد هي إحدى الأمراض التي…
أكمل القراءة » -
الأمراض الوراثية
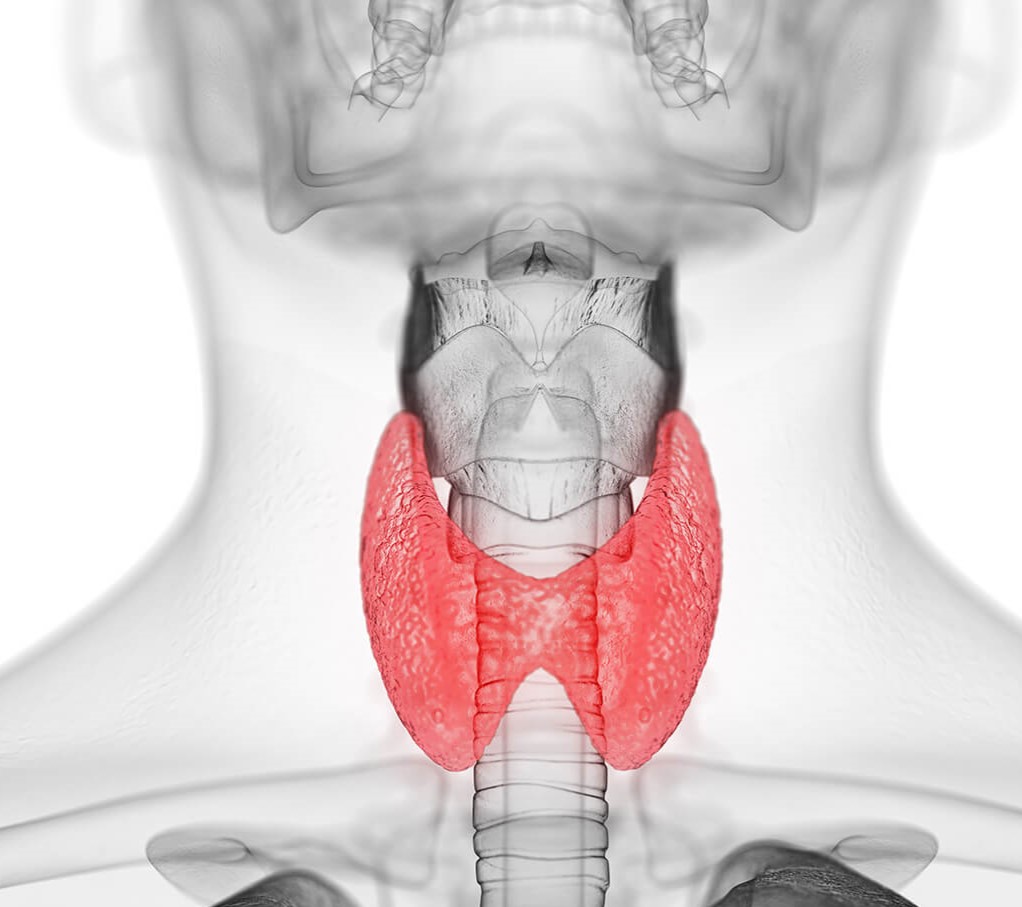
متلازمة بندرد
متلازمة بندرد Pendred syndrome في عام 1896م وصف الدكتور البريطاني فوفان بندرد عائلة ايرلندية لها طفلان لديهم صمم منذ الولادة…
أكمل القراءة »