
-
المتلازمات
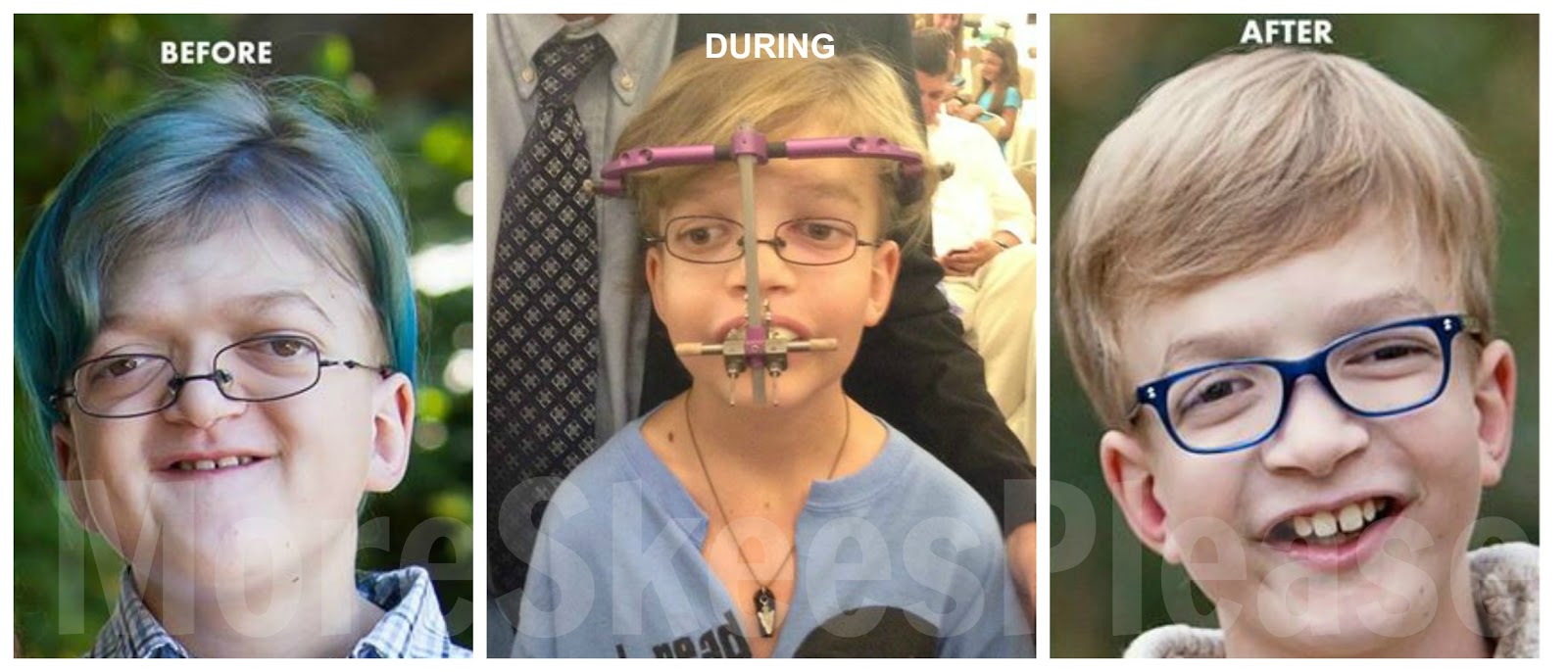
apert syndrome
متلازمة ابير Apert syndrome متلازمة ابير نوع من أنواع الانغلاق المبكر بدرجة معقدة سميت باسم دكتور فرنسي…
أكمل القراءة » -
المتلازمات

متلازمة ابرت
متلازمة ابيرت Apert syndrome متلازمة ابيرت نوع من أنواع الانغلاق المبكر بدرجة معقدة سميت باسم دكتور فرنسي وصف…
أكمل القراءة » -
متلازمة كورنيليا دي لانج
متلازمة كورنيليا دي لانج Cornelia de Lange syndrome اسماء مترادفة – متلازمة الشعر الكثيف Bushy Syndrome – اقزام امستردام. Amsterdam…
أكمل القراءة » -
مواد حملة اليوم العالمي للوقاية من التشوهات الخلقية
حملة اليوم العالمي للوقاية من المشاكل الخلقية 3 مارس 2018 هيدر لصفحات الشبكات الاجتماعية 1بوستر الحملة…
أكمل القراءة » -
حملة اليوم العالمي للوقاية من المشاكل الخلقية2018
قيل في الاثر: درهم وقاية خير من قنطارعلاج من الملاحظ ان الامراض بشكل عام مع التطورالمدني في ازدياد و الكثير…
أكمل القراءة » -
الوراثة و متلازمة جوبرت
الوراثة و متلازمة جوبرت متلازمة جوبرت هو مرض من أمراض الوراثة المتنحية. لمعرفة معلومات عامة عن المرض اقرأ هذه الصفحة.…
أكمل القراءة » -
إحصائيات عن حمض الفوليك قبل الحمل
الصلب المشقوق و حمض الفوليك لقد ثبت علمياً و بعدة بحوث علمية متكررة أن حمض الفوليك أو…
أكمل القراءة » -
campaign

أسئلة شائعة ومهمة عن حمض الفوليك
أسئلة شائعة ومهمة عن حمض الفوليك حمض الفوليك هو أحد الفيتامينات التي أثبتت التجارب العملية أهميته في تخفيض نسبة الإعاقة…
أكمل القراءة » -
كيف أنسق حملة توعية لأهمية حمض الفوليك قبل الحمل؟
كيف أنسق حملة توعية لأهمية حمض الفوليك قبل الحمل؟ كيف أنسق حملة توعية لأهمية حمض الفوليك قبل الحمل؟…
أكمل القراءة » -
ما هو يوم التوعية بأهمية حمض الفوليك قبل الحمل؟
ما هو يوم التوعية بأهمية حمض الفوليك قبل الحمل؟ هو يوم توعوي سنوي یقام في شهر يناير من كل…
أكمل القراءة » -
إحصائيات عن الفحص قبل الزواج
الفحص قبل الزواج في السعودية تم تطبيق برنامج الفحص قبل الزواج الالزامي في المملكة العربية السعودية عام … و هو…
أكمل القراءة » -
أسئلة شائعة ومهمة عن الفحص قبل الزواج
أسئلة شائعة ومهمة عن الفحص قبل الزواج ما هو الفحص الطبي قبل الزواج ؟ هو فحص طبي للمقبلين…
أكمل القراءة » -
كيف أنسق حملة توعية للفحص قبل الزواج؟
كيف أنسق حملة توعية للفحص قبل الزواج؟ كيف أنسق حملة توعية لأهمية الفحص قبل الزواج؟ يتم الاحتفال بيوم التوعية…
أكمل القراءة » -
campaign

ما هو يوم التوعية بأهمية الفحص قبل الزواج؟
ما هو يوم التوعية بأهمية الفحص قبل الزواج؟ هو يوم توعوي سنوي یقام في شهر ##### من كل عام، وقد…
أكمل القراءة » -
متلازمة القلب والوجه والبشرة
متلازمة القلب والوجه والبشرة cardiofaciocutaneous syndrome (CFC) هي اضطراب وراثي نادر صنف لأول مره عام 1986م، يصيب بنسب متفاوتة الذكور…
أكمل القراءة » -
متلازمة لارون
متلازمة لارون Laron syndrome وصف دكتور زلي لارون طبيب أمراض الغدد الصماء عام 1966 ميلادية أطفال لديهم قصر شديد في…
أكمل القراءة » -
متلازمة سانجد سقطي
متلازمة سانجد سقطي Sanjad Sakati syndrome في عام 1988 نشر مستشفى الملك فيصل التخصصي في الرياض ،باسم د. سنجد و…
أكمل القراءة » -
متلازمة فايفر
متلازمة فايفر Pfeiffer Syndrome ما هي متلازمة فايفر تعتبر متلازمة فايفر حاله مرضية نادرة مرتبطة بكل من اتحاد بالانغلاق الدروز…
أكمل القراءة » -
أمراض الاعصاب

ضمور العضلات الشوكي
ضمور العضلات الشوكي Spinal Muscular Atrophy ما هو مرض ضمور العضلات الشوكي دعونا نوضح بعض الشيء عن تسمية هذا المرض.فاصل…
أكمل القراءة » -
متلازمة برادر ولي
متلازمة برادر ولي Prader Willi Syndrome متلازمه برادرويلي هي حاله من الحالات الوراثيه التي تؤثر على كل جزء من الجسم.…
أكمل القراءة » -
متلازمة تيرنر
متلازمة تيرنر Turner syndrome في عام 1938 لاحظ الدكتور هنري تيرنر أن مجموعة من الفتيات لديهن أشباه متقاربة فبالإضافة إلى…
أكمل القراءة » -
متلازمة وليامز
متلازمة وليامز Williams syndrome لاحظ وليامز (وهو اختصاصي بأمراض القلب من نيوزيلندا) عام 1961 أن فئة صـغيرة من مرضاه الأطفال…
أكمل القراءة » -
متلازمة مارفان
متلازمة مارفان marfan syndrome من الأمور الصعبة التي يوجهها الأطباء هو معرفة ما إذا كان الشخص الطويل مصابا…
أكمل القراءة » -
متلازمة نونان
متلازمة نونان Noonan syndrome خرجت الدكتورة –جاكولين نونان في كلية الطب في بوسطن عام 1956م،وبدأت كأول طبيبة لأمراض القلب للأطفال…
أكمل القراءة » -
متلازمة بيكوث ويدمان
متلازمة بيكوث ويدمان Beckwith Wiedemann وصف الدكتور الألماني بكوث عام 2222 والدكتور ودمان عام 3333 عدة أطفال لديهم أعراض متشابهة…
أكمل القراءة » -
انواع حثل العضلات الخلقي
حثل العضلات الخلقي (Congenital muscular dystrophy (CMD مصطلح “حثل العضلات الخلقي”(CMD) هو في الحقيقة اسم لمجموعة من حثل العضلات الذي…
أكمل القراءة » -
شاركوت ماري توث
شاركوت ماري توث Charcot–Marie–Tooth disease التعريف مرض شاركوت ماري توث هوعبارة عن مجموعة من الإضطرابات الوراثية العصبية الحسية الحركية التي…
أكمل القراءة » -
انواع حثل الحزام و الأطراف العضلي
انواع حثل الحزام و الأطراف العضلي ( LGMD ) حثل عضلات الحزام و الأطراف هو في الحقيقة ليس مرض…
أكمل القراءة » -
حثل العضلات الميوتوني
حثل العضلات الميوتوني ماهو مرض حثل العضلات هو شكل من أشكال الحثل العضلي الذي يؤثر على العضلات والعديد من الأجهزة…
أكمل القراءة » -
رنح فريدريك (فريدريك أتاكسيا)
رنح فريدريك (فريدريك أتاكسيا) Friedrich Ataxia (fa) التعريف فريدريك أتاكسيا هو إضطراب عصبي يسبب ضررا بالأعصاب الطرفية ، والتي تحمل…
أكمل القراءة » -
حثل العضلات القصيا
حثل العضلات القصيا Distal muscular dystrophy عرف لأول مره عام 1902. حثل العضلات القصيا(DD)،أو العضل الطرفي وهو اسم لمجموعة من…
أكمل القراءة » -
حثل العضلات العيني البلعومي
حثل العضلات العيني البلعومي Oculopharyngeal muscular dystrophy مرض حثل العضلات العيني البلعومي(OPMD) هو ضعف في العضلات التي تتحكم بجفون العين(يؤدي…
أكمل القراءة » -
العنايه بالجهاز التنفسي لمرضى الاعصاب وخاصه الدوشين
العنايه بالجهاز التنفسي لمرضى الاعصاب وخاصه الدوشين صحة الجهاز التنفسي هي قضية حيوية بالنسبة للأطفال والبالغين الذين يعانون من أمراض…
أكمل القراءة » -
ما الفرق بين حثل العضلات و ضمور العضلات ؟
ما الفرق بين حثل العضلات و ضمور العضلات ؟ هل الحثل العضلي له علاج ولا متل ضمور العضلات ليس له…
أكمل القراءة » -
حملة التوعية بالمشاكل الخلقية 2015
حملة التوعية بالمشاكل الخلقية 3 مارس 2015 النسخة المترجمة بالإحصائيات العالمية بوستر راعي رابط ملف pdf عالي الجودة للطباعة…
أكمل القراءة » -
متلازمة وردينبيرج
متلازمة وردينبيرج Waardenburg Syndrome يرجع اسم هذه المتلازمة إلى طبيب العيون الهولندي الدكتور بطرس جوهانز وردينبيرج (Dr. Petrus Johannes…
أكمل القراءة » -
متلازمة جولدنهار
متلازمة جولدنهار Goldenhar syndrome لهذه المتلازمة عدة أسماء .فيطلق عليها أيضا سلسلة(متلازمة) العين و الأذن و الفقرات(Oculo-Auriculo-Vertebrali Spectrum) كما…
أكمل القراءة » -
متلازمة ستكلر
متلازمة ستكلر Stickler syndrome و صف الدكتور ستكلر عام 1965م خمسة أجيال من أسرة و واحدة لديها ضعف سمع…
أكمل القراءة » -
متلازمة الخيشوم والأذن والكلية
متلازمة الخيشوم والأذن والكلية Branchio-oto-renal syndrome و صف الدكتور ملنيك هذه المتلازمة في عام 1975…
أكمل القراءة » -
متلازمة كروموسوم اكس الهش
متلازمة كروموسوم اكس الهش Fragile x syndrome قد أشارت العديد من الأبحاث الجديدة منها والقديمة أن متلازمة كروموسوم اكس المكسور…
أكمل القراءة » -
متلازمة اُوشر
متلازمة اُوشر Usher Syndrome يصيب هذا المرض جميع الشعوب و لكن الدكتور البرشت فون جريفي(. Albrecht Von Graefe) وصفة…
أكمل القراءة » -
شق الشفة و شق الحنك
شق الشفة و شق الحنك المرأة إذا رأت أرنبا فإن طفلها سيولد بشفة أرنبية!! في القديم كان هناك نظرة…
أكمل القراءة » -
الخلع الوركي الولادي
الخلع الوركي الولادي يعتقد الكثير خطاءً أن سبب خلع الورك الولادي لحديثي الولادة ناتج عن الآلات المستخدمة في التوليد أثناء…
أكمل القراءة » -
مقدمة عن المشاكل الخلقية
المشاكل الخلقية هي عبارة عن تخلق غير طبيعي في احد أعضاء الجسم أو الأنسجة في مرحلة تخلق الجنين. وعادة ما…
أكمل القراءة » -
مرض الحثل العضلي الدوشيني
مرض الحثل العضلي الدوشيني Duchenne Muscular Dystrophy عرّف مرض الحِثل العضلي بالاضطراب الجيني الذي يقوم تدريجيا بإضعاف عضلات الجسم. وسبب…
أكمل القراءة » -
الحثل العضلي الولادي
الحثل العضلي الولادي Congenital Muscular Dystrophy ما هو الحثل العضلي الولادي ؟ الحثل العضلي الولادي عباره عن مجموعه من الامراض…
أكمل القراءة » -
مرض الحثل العضلي
مقدمة عن مرض الحثل العضلي Muscular Dystrophy مرض الحِثل العضلي(Muscular Dystrophy) و يرمز له بالرمز MD هو احد امراض…
أكمل القراءة » -
مرض التليف العصبي
مرض التليف العصبي Neurofibromatosis هناك نوعان من التليف العصبي النوع الأول والنوع الثاني ومع أن لهم نفس المسمى إلا أنهما…
أكمل القراءة » -
التصلب الدرني الوراثي
التصلب الدرني الوراثي Tuberous Sclerosis التصلب الدرني هو خلل وراثي ينتقل لثلث الحالات المصابة عن طريق الوالدين اما باقي الحالات…
أكمل القراءة » -
متلازمة استطالة كيو تي
متلازمة استطالة كيو تي Long QT syndrome ماهي متلازمة استطالة كيو تي؟ إن متلازمة استطالة كيو تي هي مجموعة من…
أكمل القراءة » -
متلازمة باتاو
متلازمة باتاو كروموسوم 13 الثلاثي Trisomy 13 Syndrome ما هي متلازمة كروموسوم 13 الثلاثي متلازمة كروموسوم 13 الثلاثي (ويعرف…
أكمل القراءة » -
متلازمة إدوارد
Edward syndrome نزل متلازمة ادورد كملف بي دي اف للطباعة متلازمة إدوارد متلازمة إدوارد هي إحدى الأمراض التي…
أكمل القراءة » -
متلازمة تشارج
متلازمة تشارج CHARGE Syndrome في عام 1979 نشر الدكتور براين هول في مجلة طبية للأطفال و صف 17…
أكمل القراءة » -
الأمراض الوراثية
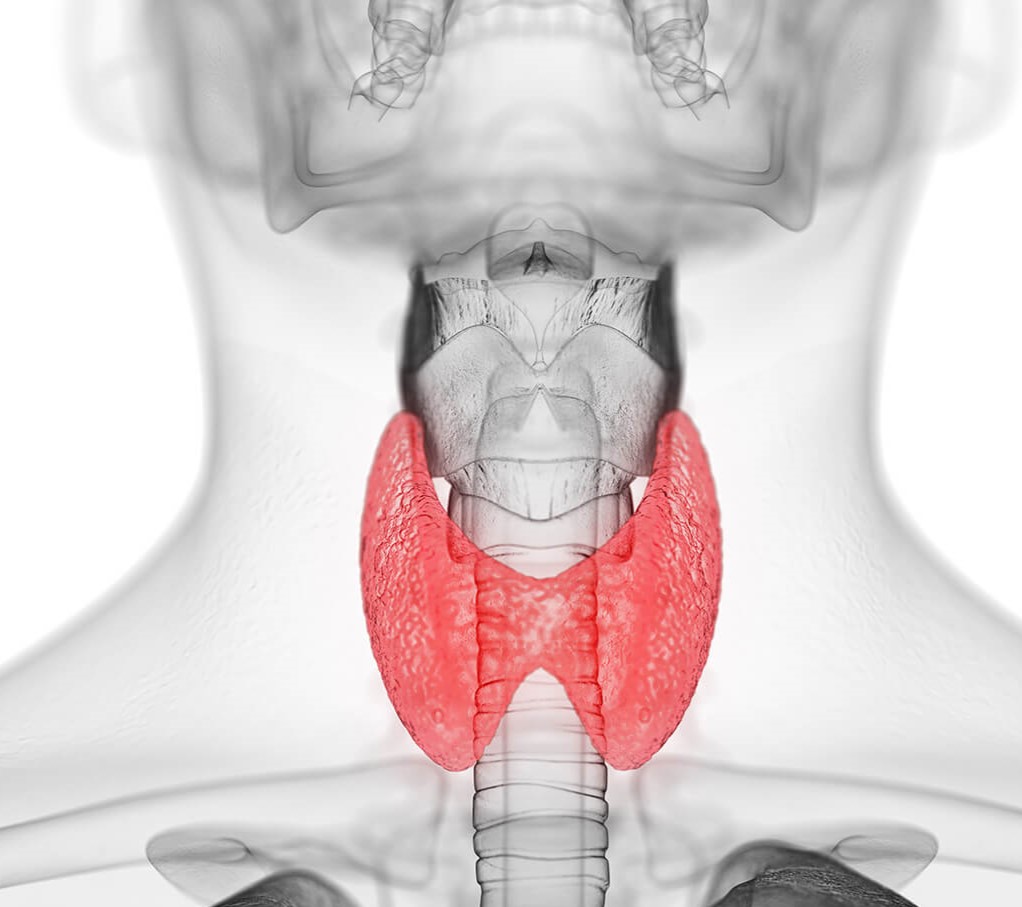
متلازمة بندرد
متلازمة بندرد Pendred syndrome في عام 1896م وصف الدكتور البريطاني فوفان بندرد عائلة ايرلندية لها طفلان لديهم صمم منذ الولادة…
أكمل القراءة » -
متلازمة ميكل جروبر
متلازمة ميكل جروبر MECKEL-GRUBER SYNDROME متلازمة ميكل جروبر هي مرض وراثي نتيجة لخلل في احد الجينات ينتقل بالوراثة المتنحية…
أكمل القراءة » -
متلازمة جوبيرت
متلازمة جوبيرت Joubert Syndrome متلازمة جوبيرت هي اضطراب أو خلل في جزء من الدماغ يسمى الدودة المخيخية (جسر المخيخ) …
أكمل القراءة » -
برودكاست الوراثة الطبية
برودكاست الوراثة على الواتس أب WhatsApp أهلا وسهلاً بكم معنا في برودكاست الوراثة الطبية في هذه الصفحة سنشارككم بالرسائل التي…
أكمل القراءة » -
مجموعة الدعم للمتلازمات الوراثية والخلقية
مرحبا بكم معنا في مجموعة الدعم الأسري للمتلازمات الوراثية و الخلقية التي تنطلق من موقع ومننتديات الوراثة الطبية تحت رعاية…
أكمل القراءة » -
دعوة للمشاركة!
يحرص موقع الوراثة الطبية على استقطاب نخبة من الأسر والمرضى الذين لهم طموحات ونظره عميقة في مجال الدعم الذاتي الأسري…
أكمل القراءة » -
القيم التي نعتز بها
القيم التي نعتز بها الدعم الأسري المستمر الغير ربحي التميز في العمل الجماعي التطوعي الفعال التميز في أساليب التوعية دقة…
أكمل القراءة » -
الرؤية والرسالة
رؤيتنا تحقيق الريادة والتميز عربيا و إلكترونيا في التوعية و الدعم الأسري في مجال الأمراض الوراثية والخلقية. رسالتنا أول و…
أكمل القراءة »