
المتلازمات
-
apert syndrome
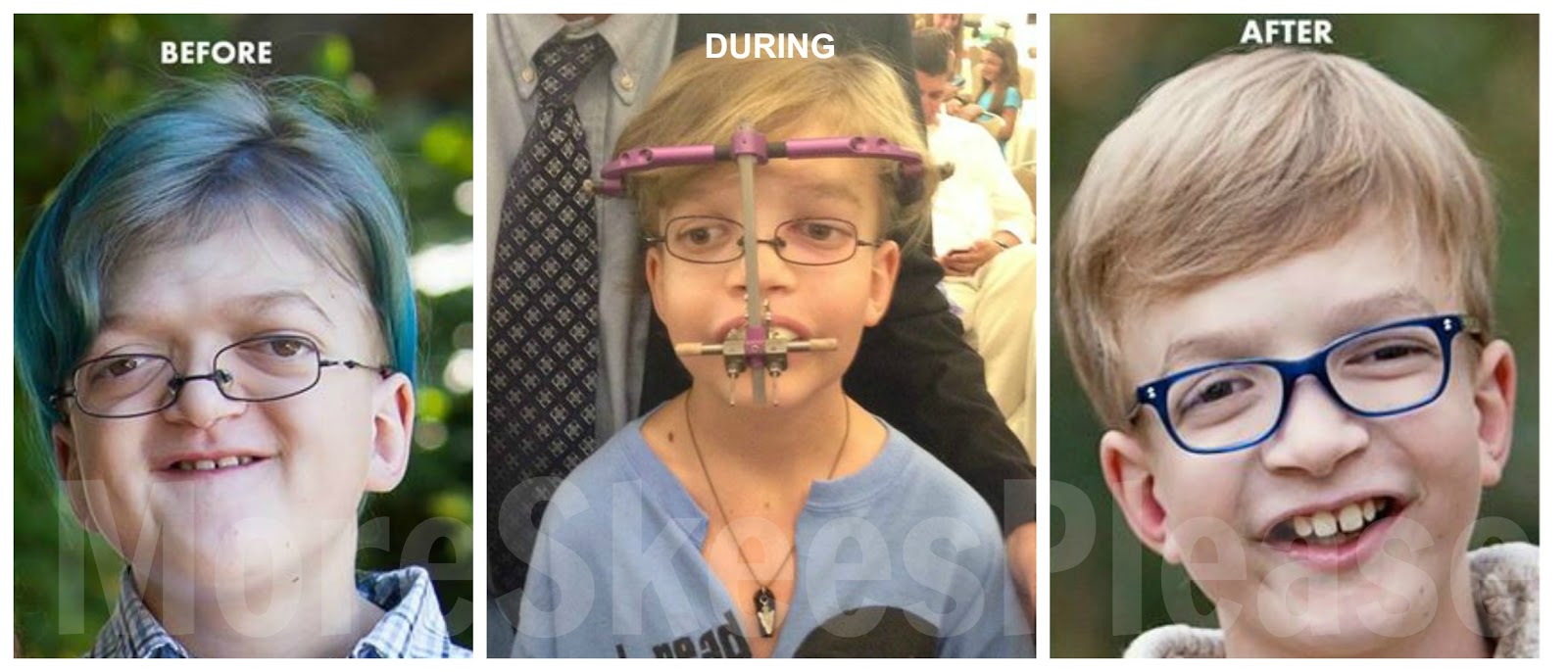
متلازمة ابير Apert syndrome متلازمة ابير نوع من أنواع الانغلاق المبكر بدرجة معقدة سميت باسم دكتور فرنسي…
أكمل القراءة » -

متلازمة ابرت
متلازمة ابيرت Apert syndrome متلازمة ابيرت نوع من أنواع الانغلاق المبكر بدرجة معقدة سميت باسم دكتور فرنسي وصف…
أكمل القراءة » -
متلازمة كورنيليا دي لانج
متلازمة كورنيليا دي لانج Cornelia de Lange syndrome اسماء مترادفة – متلازمة الشعر الكثيف Bushy Syndrome – اقزام امستردام. Amsterdam…
أكمل القراءة » -
متلازمة القلب والوجه والبشرة
متلازمة القلب والوجه والبشرة cardiofaciocutaneous syndrome (CFC) هي اضطراب وراثي نادر صنف لأول مره عام 1986م، يصيب بنسب متفاوتة الذكور…
أكمل القراءة » -
متلازمة لارون
متلازمة لارون Laron syndrome وصف دكتور زلي لارون طبيب أمراض الغدد الصماء عام 1966 ميلادية أطفال لديهم قصر شديد في…
أكمل القراءة » -
متلازمة سانجد سقطي
متلازمة سانجد سقطي Sanjad Sakati syndrome في عام 1988 نشر مستشفى الملك فيصل التخصصي في الرياض ،باسم د. سنجد و…
أكمل القراءة » -
متلازمة فايفر
متلازمة فايفر Pfeiffer Syndrome ما هي متلازمة فايفر تعتبر متلازمة فايفر حاله مرضية نادرة مرتبطة بكل من اتحاد بالانغلاق الدروز…
أكمل القراءة » -
متلازمة برادر ولي
متلازمة برادر ولي Prader Willi Syndrome متلازمه برادرويلي هي حاله من الحالات الوراثيه التي تؤثر على كل جزء من الجسم.…
أكمل القراءة » -
متلازمة تيرنر
متلازمة تيرنر Turner syndrome في عام 1938 لاحظ الدكتور هنري تيرنر أن مجموعة من الفتيات لديهن أشباه متقاربة فبالإضافة إلى…
أكمل القراءة » -
متلازمة وليامز
متلازمة وليامز Williams syndrome لاحظ وليامز (وهو اختصاصي بأمراض القلب من نيوزيلندا) عام 1961 أن فئة صـغيرة من مرضاه الأطفال…
أكمل القراءة » -
متلازمة مارفان
متلازمة مارفان marfan syndrome من الأمور الصعبة التي يوجهها الأطباء هو معرفة ما إذا كان الشخص الطويل مصابا…
أكمل القراءة » -
متلازمة نونان
متلازمة نونان Noonan syndrome خرجت الدكتورة –جاكولين نونان في كلية الطب في بوسطن عام 1956م،وبدأت كأول طبيبة لأمراض القلب للأطفال…
أكمل القراءة » -
متلازمة بيكوث ويدمان
متلازمة بيكوث ويدمان Beckwith Wiedemann وصف الدكتور الألماني بكوث عام 2222 والدكتور ودمان عام 3333 عدة أطفال لديهم أعراض متشابهة…
أكمل القراءة » -
متلازمة وردينبيرج
متلازمة وردينبيرج Waardenburg Syndrome يرجع اسم هذه المتلازمة إلى طبيب العيون الهولندي الدكتور بطرس جوهانز وردينبيرج (Dr. Petrus Johannes…
أكمل القراءة » -
متلازمة جولدنهار
متلازمة جولدنهار Goldenhar syndrome لهذه المتلازمة عدة أسماء .فيطلق عليها أيضا سلسلة(متلازمة) العين و الأذن و الفقرات(Oculo-Auriculo-Vertebrali Spectrum) كما…
أكمل القراءة » -
متلازمة ستكلر
متلازمة ستكلر Stickler syndrome و صف الدكتور ستكلر عام 1965م خمسة أجيال من أسرة و واحدة لديها ضعف سمع…
أكمل القراءة » -
متلازمة الخيشوم والأذن والكلية
متلازمة الخيشوم والأذن والكلية Branchio-oto-renal syndrome و صف الدكتور ملنيك هذه المتلازمة في عام 1975…
أكمل القراءة » -
متلازمة كروموسوم اكس الهش
متلازمة كروموسوم اكس الهش Fragile x syndrome قد أشارت العديد من الأبحاث الجديدة منها والقديمة أن متلازمة كروموسوم اكس المكسور…
أكمل القراءة » -
متلازمة استطالة كيو تي
متلازمة استطالة كيو تي Long QT syndrome ماهي متلازمة استطالة كيو تي؟ إن متلازمة استطالة كيو تي هي مجموعة من…
أكمل القراءة » -
متلازمة باتاو
متلازمة باتاو كروموسوم 13 الثلاثي Trisomy 13 Syndrome ما هي متلازمة كروموسوم 13 الثلاثي متلازمة كروموسوم 13 الثلاثي (ويعرف…
أكمل القراءة » -
متلازمة إدوارد
Edward syndrome نزل متلازمة ادورد كملف بي دي اف للطباعة متلازمة إدوارد متلازمة إدوارد هي إحدى الأمراض التي…
أكمل القراءة » -
متلازمة ميكل جروبر
متلازمة ميكل جروبر MECKEL-GRUBER SYNDROME متلازمة ميكل جروبر هي مرض وراثي نتيجة لخلل في احد الجينات ينتقل بالوراثة المتنحية…
أكمل القراءة »